Project Summary
Long-acting injectables (LAI) have been developed to offer prolonged drug release and thus improve treatment adherence. Approximately 30 LAI drug products are currently approved by the FDA. However, only one of these has an approved generic formulation because the long terminal half-life and high inter-individual variability result in risky bioequivalence trials with long duration and low power. Here we propose a novel BE design combined with a model-integrated approach to reduce the duration of bioequivalence trials for LAIs.
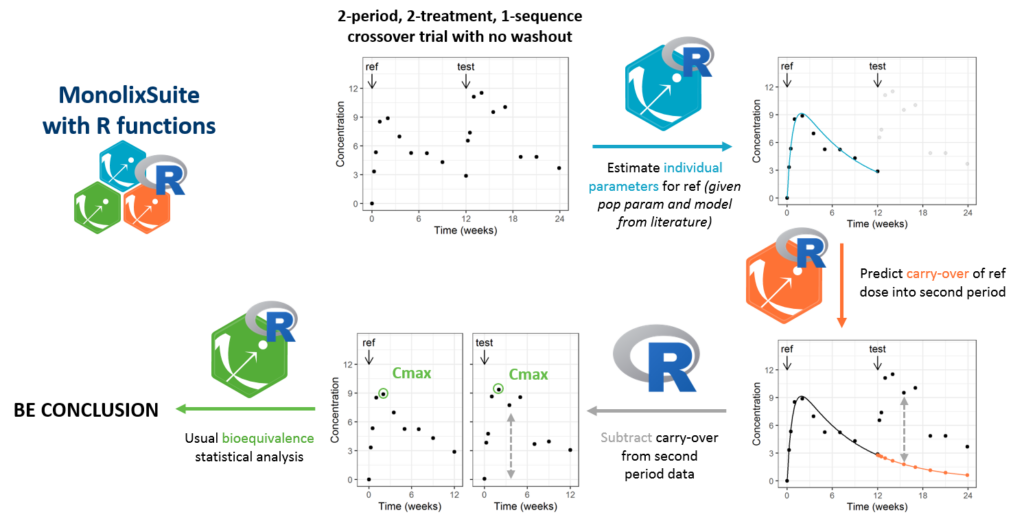
The novel design we propose is a 2-treatment, 2-period, 1-sequence “reduced crossover” with no or limited washout. The data of the second period takes into account the second dose and the carryover from the first dose and can thus not be directly compared to the data of the first period. Therefore, we apply a model-based correction of the data of the second period. To do so, individual parameters are estimated on the data of the first period (reference formulation, for which a population model is usually available) and used to predict the carryover concentration into the second period. The predicted carryover concentration is then subtracted from the second period data before usual BE analysis. The method is applicable in the case of a single dose design (i.e on healthy volunteers), and linear PK (i.e superposition principle applies).
The procedure is exemplified with Buprenorphine LAI, using a published model. To be valuable, the proposed procedure needs to have a properly controlled type I error (probability of wrongly concluding bioequivalence, i.e patient risk) and a sufficiently high power (probability of correctly concluding bioequivalence). To calculate the empirical type I error and power, many BE trials with the proposed “reduced crossover” design are simulated under the null hypothesis (no bioequivalence) or the alternative hypothesis (bioequivalence) and submitted to the analysis procedure. For comparison, power and type I error is also calculated for classical crossovers with washout period and parallel designs.
For the reduced crossover design with an inter-dose interval of 4 months, the type I error is properly controlled, and the power is above 90% with 30 individuals. This reduced crossover design is thus three times shorter than the traditional crossover and requires a 10 times smaller sample size compared to a parallel trial.
The proposed model-integrated reduced crossover bioequivalence design and the associated analysis procedure is shown to provide a high power for a reasonable study duration and sample size, with a properly controlled type I error. It alleviates the current challenges for the development of LAI generics and represents a hope for the community. The analysis procedure is implemented as an R script, relying on the MonolixSuite. It can easily be applied to other LAIs.